Ketonuria ana ing urine awak ketone, yaiku aseton
Mekanisme ketonuria.
Ing wong sing sehat, karbohidrat, lipid, lan sawetara protein disoksidasi dadi karbon dioksida lan banyu. Ing sawetara kahanan patologis, utamane diabetes, produksi insulin nyuda. Ing ati, cadangan glikogen dikurangi, akeh jaringan awak sing keluwen energi. Ing kahanan kasebut, proses oksidasi protein lan lipid ing ati diaktifake, nanging kurang glikogen ndadékaké oksidasi sing ora lengkap lan akumulasi ing getih produk metabolisme lipid lan protein sing ora disoksidasi - awak keton. Amarga klumpukan awak keton ing getih (ketonemia), pH getih pindhah menyang sisih asam. Kahanan iki diarani acidosis. Urine pasien kaya iki duwe reaksi asam sing cetha lan mambu aseton.
Keluwen, panggunaan panganan protein lan lemak utamane, pengecualian karbohidrat saka diet nyebabake tambah kabentuk awak keton lan ekskresi ing urin.
Ing umur wiwitan, ketonuria luwih umum tinimbang karo wong diwasa lan ora duwe teges klinis. Fenomena iki pancen narik kawigaten karo dokter anak karo "mutetoemis" sing ana gandhengane karo kasalahan ing diet.
Ketonuria ing wektu pasca operasi amarga karusakan protein amarga trauma bedah.
Ing wong diwasa, ketonuria dumadi ing macem-macem diabetes lan menehi nilai diagnosa sing gedhe. Ing bocah-bocah, bisa uga macem-macem penyakit, amarga metabolisme karbohidrat. Dadi, sanajan kesalahan cilik ing diet, utamane ing ngarsane infeksi akut, kegirangan gemeter, overwork, lsp. bisa nyebabake ketosis. Ketonuria nalika bocah cilik bisa diamati karo toksikosis, gangguan gastrointestinal sing berpanjangan, disentri lan penyakit liyane. Ing bayi sing nembe bayi, kenaikan keton cipratan meh asring disebabake dening kekurangan gizi. Ketonuria, sing diamati ing penyakit infèksius - mriyang abang, flu, meningitis tuberkolus lan mabuk, minangka tandha sekunder, tetep lan ora duwe nilai diagnosa sing gedhe.
Ketonuria kanggo diabetes berkembang amarga tambah ketogenesis lan ketolysis mboten saget. Ketogenesis tambah nyebabake mobilisasi lemak tambah saka jaringan adipose, nyuda pembentukan oxalacetate ing siklus Krebs, lan nyuda biosintesis asam lemak. Ing diabetes sing parah, disertai kerusakan ing jaringan ginjel (papan kanggo pecah keton), pelanggaran tambahan ketolysis ana.
Anane glukosuria ing ngarsane ketonuria ora kalebu diabetes.
Biasane, awak keton dibentuk kanthi sithik saka produk akhir metabolisme karbohidrat lan lipid - acetyl-CoA (C2-bodies) liwat acetoacetyl-CoA lan meh ora netralisasi.
Ing diabetes mellitus, mobilisasi lipid tambah kanthi pembentukan jumlah acetyl CoA (C2awak). ing wektu sing padha, amarga nglanggar metabolisme karbohidrat, nyuda pembentukan oxalacetate, sing kalebu C2-buwang kalebu ing siklus Krebs lan bakar nganti karbon dioksida lan banyu. Kajaba iku, minangka akibat saka paningkatan lipid, biosintesis sabalikna C diblokir.2awak dadi asam lemak. Mangkono, jumlah C nglumpukake2Sapa wae, sing ndadékaké produksi CoA, lan asam asetat, acetoacetic lan beta-hydroxybutyric acid, sing diekskresi ing urin.
Badan ketone
Iki minangka produk bosok penengah sing dibentuk sajrone fungsi ati. Biasane, metabolisme mbabarake badan keton dadi luwih rusak.
Resep glukosa awak ditemokake ing gajih awak, mulane ketonuria kekurangan karbohidrat. Badan ketone minangka panyedhiya energi sing apik banget. Ing iki, malah asam lemak ketinggalan. Mula, nalika otak utawa jantung kekurangan asam fosfat, awak langsung ngasilake awak keton kanthi cepet.
Apa alasan
Ketonuria yaiku isi urin awak ketone ing ndhuwur normal.
Ing awak manungsa sing sehat, metabolisme wis seimbang. Apa sing bisa nyebabake ketonuria?

- Keuntungan protein lan lemak ing panganan. Amarga nyuda karbohidrat ing panganan, sel kurang gizi. Ngatasi latar mburi iki, ketonuria berkembang. Iki minangka reaksi awak kanggo ora seimbang ing diet.
- Diet lan penyalahgunaan keluwen bisa nyebabake munculé keton awak ing cipratan. Kesalahan ing pilihan diet nyebabake lemak cepet banget, dene jumlah enzim saya tambah. Ketonuria uga munculé aseton ing getih. Biasane, yen wong keluwen luwih saka enem dina, isi awak keton ing awak manungsa mundhak normal.
- Anestesi kanggo operasi.
- Diabetes mellitus. Ing kasus iki, ketonuria katon saka wektu. Badan ketone mung dudu panyebab penyakit kasebut. Ing diabetes mellitus, tambahan karbohidrat lan insulin diwènèhaké.
- Dehidrasi. Iki dumadi nalika awak ngalami overheats utawa komplikasi diabetes.
- Penyakit jantung utawa penyakit infèksius abot (mis., Disentri).
- Perkembangan tumors ing saluran pencernaan.
- Gangguan ing pankreas.
- Keracunan dening alkohol utawa dening senyawa kimia kaya fosfor, timbal.
- Kerusakan sistem saraf pusat, kegirangan gemeter.
Elinga, ambune aseton nalika ambegan utawa urinating minangka sinyal kanggo ndeleng dhokter. Sampeyan uga dadi sebab kanggo ngganti gaya urip lan keseimbangan nutrisi.
Manut marang anak
Klinik kasebut asring ditliti yen bocah kasebut muntah, mambu aseton. Utawa mambu kaya mengkono muncul ing cipratan. Lan sanajan iku tandha ketonuria, mesthine ora ana gejala sing lara.

Asring, kabeh diterangake dening sistem metabolisme sing ora sampurna. Penyebab ketonuria ing bocah-bocah yaiku kondhisi energi sing dibuwang. Biasane dumadi nalika:
- garan emosi
- tambah semangat gedhe,
- diet sing ora seimbang
- kadhemen
Kasunyatane yaiku yen awak bocah ora duwe toko glikogen sing akeh, mula, lemak pecah aktif dumadi lan tanda-tanda ketonuria diamati.
Kandhutan
Sajrone meteng sangang wulan, wanita asring kudu njupuk tes urin. Iki pancen prelu ora nyasar sethithik penyimpangan saka norma ing awak sajrone wektu gestasi. Sawise kabeh, kabeh organ duwe beban gedhe. Apa tegese awak keton ditemokake ing urin?
Biasane konten cilik kasebut minangka norma. Analisis paling gampang bakal ngliwati tes cepet. Sampeyan kudu nyuda jalur tes ing cipratan.
Tes negatif dianggep normal sajrone meteng utawa jumlah keton minimal. Yen nilai uji coba saka 15 nganti 160 mg / dl - iki dadi prihatin.
Sajrone meteng, ketonuria minangka gejala sing nguwatirake. Katon karo toksikosis awal. Keracunan awak, lan saka iku janin kanthi aseton, ngrampungake meteng.

Yen wanita meteng duwe kegagalan hormon sing serius, mula ana risiko.
Tambah saka tingkat keton ing cipratan bayi sing anyar amarga kekurangan pangan utawa kesalahan nutrisi.
Gejala Ketonuria
Yen tingkat keton ing awak saya tambah, mula ketonuria bakal katon dhewe:
- kesel banget,
- mambu aseton saka tutuk,
- ambu ora enak aseton
- analisa bakal nuduhake tingkat getih putih ing getih,
- tes getih biokimia bakal nuduhake konten glukosa sing sithik,
Yen ketonuria provokasi ucul saka getih aseton, bisa uga ana krisis aseton.
Tambah ing acidity sing signifikan ing sel bisa ngrusak organ internal. Ing kasus iki, reaksi proteksi diluncurake - muntah.
Gejala ketonuria ing bocah-bocah:
- keluhan weteng
- keluhan sirah
- bocah wis kesel, lesu,
- keluhan mual
- mutah
- suhu suhu 39 ° C,
- Nolak pangan,
- mambu kaya aseton
- nggedhekake ati
Apa ketonuria bisa dideteksi?
Mung analisa kimia sing bisa ndeteksi tingkat keton ing awak. Laboratorium bakal langsung netepake norma awak keton sing ana.
Ing obat modern, ketonuria dideteksi:
- Uji Lange,
- Tes hukum
- Contoh Lestrade,
- conto diowahi Rothera,
- tes cepet

Tes sing cepet, mesthi wae. Tumindak kasebut adhedhasar reaksi kimia, asil sing katon meh langsung. Siji kudu mung nyelehake jalur tes ing urin utawa selehake ing tablet test. Ing kasus reaksi positif, tes dadi ungu. Padhange warna ngidini sampeyan ngadili kanthi skala warna khusus babagan kepiye norma awak keton ngluwihi.
Klasifikasi Internasional Penyakit
Klasifikasi Statistik Internasional Penyakit lan Masalah Kesehatan sing Terkait (ICD) minangka pitunjuk referensi sing bisa dibandhingake kanthi internasional. Kanthi menehi pitulung, macem-macem pendekatan metodologis digabungake. Statistik penyakit dikumpulake lan diklasifikasikake. Saben sepuluh taun, IBC diteliti dening komisi Organisasi Kesehatan Dunia. Supaya klasifikasi lan nganalisa sakabehing asil sing diklumpukake saka sudut pandang statistik, jarene diterjemahake menyang kode alphanumeric. Ing karya kasebut, ICD sing dikembangake digunakake. Dina iki cocog karo ICD-10. Ngemot 22 kelas (bagian).
Miturut direktori ICD, ketonuria duwe kode R82.4.
Nyegah lan Diet Ketonuria
Kanggo nyegah ketonuria, perlu:
- mangan tengen
- mimpin gaya urip sehat:
- sabisa bisa mlebu hawa seger,
- olahraga sing moderat
- Aja miwiti penyakit nemen.

Ing penyakit kronis kayata diabetes, dhokter konsultasi kanthi sistematis. Saka wektune perlu kanggo njupuk tes ekspres.
Reaksi positif nuduhake patologi ing awak. Pay manungsa waé menyang sinyal! Mréntahaké ing klinik kanthi cepet bakal mulih awak kanthi normal. Tanpa minggat saka panganan sing diwenehake dening dokter, sampeyan bisa nyepetake pemulihan.
Prinsip diet ketonuria:
- aja mangan panganan sing akeh protein lan lemak,
- intake karbohidrat biasa
- ngombe solusi banyu lan soda luwih akeh (kanthi diabetes - insulin).
Menu kasebut kudu kalebu: rebus daging sapi lan terwelu, macem-macem sup sayur, iwak rendah lemak. Sereal sing migunani tanpa minyak, sayuran lan woh. Disaranake ngombe jus liyane, omben-omben, compotes.
Ngilangi saka menu:
- daging lemu
- bumbu pedhes
- manis
- woh jeruk
- gedhang
- jamur
- panganan cepet.
Fokus perawatan
Ketonuria ora kudu dianggep minangka penyakit sing kapisah. Dheweke mung konsekuensi. Sampeyan perlu kanggo ngilangi sebab-sebab sing nyebabake. Pisanan, dibutuhake lengkap. Ketepatan diagnosis lan panyiapan penyebab ketonuria njamin perawatan sukses.
Dokter menehi sawetara tips:
- Yen sampeyan lagi kabotan, sampeyan kudu ngatur dina-dina pasa.
- Yen analisis nyiptakake kenaikan keton urin, mula tuku tes digunakake ing omah.

- Urin kudu diklumpukake kanggo nganalisa ora luwih saka patang jam sadurunge dikirim.
- Bocah kasebut bisa diombe kanthi ngombe alkali saben 10-15 menit ing bagian cilik. Arang aktif, enterosgel bakal mbantu ngilangi saluran pencernaan.
- Kanthi muntah mbesuk, migunani kanggo ngombe minuman keras. Telpon ambulans.
Cara ngobati ketonuria, ana tips ing obat rakyat.
- Kanthi mutahke, ngombe banyu mineral, kompote buah garing, larutan glukosa. Siji sendok ing sepuluh menit.
- Sijine enema ing omah. Pisanan, banyu ing suhu kamar, mengko anget, sing ditambah karo sendok teh soda.
- Ngombe: bubar 2 sudu besar 1 liter banyu. madu, tuangake jus siji linglang. Ngombe 1 tbsp. saben 15 menit.
- Resep kanggo solusi soda: larut 1 sendok teh soda ing 250 ml banyu. Ngombe 1 sendhok teh. saben 10 menit.
- Njupuk decoctions saka tanduran sing nyenengake.
- Kanggo nyepetake penghapusan racun saka awak, sithik mangan sithik. Luwih becik mangan mung krupuk.
Perawatan gumantung marang karakteristik individu ing awak. Iki kudu dikendhaleni kanthi ketat dening dokter sing nekani.
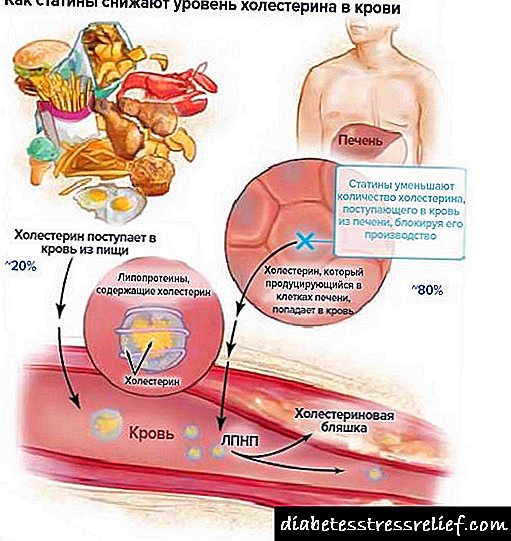
76. Kolesterol. Cara mlebu, nggunakake lan ekskresi saka awak. Kolesterol serum Biosintesis kolesterol, tahapane. Regulasi sintesis.
Kolesterol minangka steroid spesifik kanggo organisme kewan. Disintesis ing pirang-pirang jaringan manungsa, nanging sintesis utama yaiku ati. Ing ati, luwih saka 50% kolesterol disintesis, ing usus cilik - 15-20%, sisa kolesterol disintesis ing kulit, korteks adrenal, lan gonads. Kira-kira 1 g kolesterol disintesis saben dina ing awak, 300-500 mg dies ing panganan (Gambar 8-65). Kolesterol nindakake pirang-pirang fungsi: iki minangka bagean saka kabeh membran sel lan mengaruhi sifat-sifat, dadi substrat sintesis asam empedu lan hormon steroid. Prekursor ing jalur metabolis sintesis kolesterol uga dadi ubiquinone, yaiku komponen rantai pernafasan lan dolichol, sing melu sintesis glikoprotein. Amarga klompok hidroksil, kolesterol bisa mbentuk ester kanthi asam lemak. Kolesterol kolesterol Etherified ndarbeni ing getih lan disimpen kanthi jumlah sithik ing sawetara jinis sel sing nggunakake minangka landasan sintesis bahan liyane. Kolesterol lan ester kasebut minangka molekul hidrofobik, saengga diangkut getih mung minangka bagian saka macem-macem jinis obat. Bursa kolesterol iku kompleks banget - mung kanggo sintesis, udakara 100 reaksi berturut-turut kudu. Gunggunge kira-kira 300 protein beda melu metabolisme kolesterol. Gangguan metabolisme kolesterol nyebabake salah sawijining penyakit sing paling umum - aterosklerosis. Mortalitas saka efek atherosclerosis (infark miokard, stroke) nyebabake struktur kematian total. Atherosclerosis minangka "penyakit poligenik", i.e. akeh faktor sing melu ngembangake, sing paling penting yaiku keturunan. Akumulasi kolesterol ing awak ndadékaké penyakit liyane sing padha - penyakit gallstone.
A. Sintesis kolesterol lan pengaturane
Reaksi sintesis kolesterol dumadi ing sitosol sel. Iki minangka salah sawijining jalur metabolis paling dawa ing awak manungsa.
Phenylketonuria

Phenylketonuria - pelanggaran metabolisme asam amino amarga kekurangan enzim ati sing melu metabolisme phenylalanine menyang tirosin. Tanda-tanda awal phenylketonuria yaiku muntah, lesu utawa hiperaktif, mambu saka urin lan kulit, pangembangan psikomotor sing tundha, tandha-tandha pungkasan yaiku kalebu oligofrenia, wektu tundha pangembangan fisik, kombulsi, owah-owahan kulit, ekzematous, lan liya-liyane. diagnosa sakteruse kalebu tes genetik molekular, tekad konsentrasi phenylalanine getih, analisis biokimia urine, EEG, lan MRI otak. Pengobatan phenylketonuria yaiku ngetutake diet khusus.

Informasi umum
Phenylketonuria (Penyebaran Felling, oligofrenia phenylpyruvic) yaiku patologi kongenital, ditemtokake sacara genetik kanthi hidroksilasi gangguan saka phenylalanine, akumulasi asam amino lan metabolit ing cairan fisiologis lan jaringan, banjur disebabake dening karusakan sistem saraf pusat. Phenylketonuria sepisanan diterangake dening A. Felling ing taun 1934, kedadeyan kanthi frekuensi 1 kasus saben 10,000 bayi. Ing periode neonatal, phenylketonuria ora duwe kawujudan klinis, nanging intake phenylalanine kanthi pangan nyebabake manifestasi penyakit sing wis ana ing separo sepisanan, lan sabanjure nyebabake gangguan perkembangan bocah sing abot. Pramila deteksi phenylketonuria pra-gejala ing bayi yaiku tugas neonatologi, pediatrik lan genetika.
Panyebab Phenylketonuria
Phenylketonuria minangka kelainan pusing resesif autosomal. Iki tegese kanggo ngembangake tandha-tandha klinis phenylketonuria, bocah kasebut kudu menehi salah sawijining salinan gen sing salah saka wong tuwa, yaiku operator heterozygous saka gen mutant.
Paling asring, pangembangan phenylketonuria disababaké amarga mutasi ing génom sing ngétode enzim phenylalanine-4-hydroxylase sing dumunung ing lengen kromosom 12 (lokus 12q22-q24.1). Iki sing diarani jinis klasik phenylketonuria, sing nyakup 98% saka kabeh kasus penyakit. Hyperphenylalaninemia bisa nganti 30 mg% lan luwih dhuwur. Yen ora ditambani, varian phenylketonuria iki diiringi mundur mental.
Saliyane ing bentuk klasik, phenylketonuria atypical dibedakake, diterusake kanthi gejala klinis sing padha, nanging ora bisa didandani kanthi terapi diet. Iki kalebu jinis phenylketonuria II (kurang pengurangan dehidroterterin), jinis phenylketonuria III (kurang tetrahydrobiopterin) lan liyane, variasi liyane.
Kemungkinan nglairake anak kanthi phenylketonuria mundhak karo kekawin sing cedhak.
Patogenesis saka phenylketonuria
Bentuk klasik phenylketonuria adhedhasar kurang saka enzim phenylalanine-4-hidroksilase sing melu konversi phenylalanine dadi tirosin ing mitokondria hepatosit. Ing giliran, tyramine turunan tyrosine minangka bahan wiwitan kanggo sintesis catecholamines (adrenalin lan norepinephrine), lan diiodotyrosine kanggo pembentukan tiroksin. Kajaba iku, pambentukan pigmen melanin minangka metabolisme phenylalanine.
Kekurangan keturunan enzim phenylalayin-4-hydroxylase ing phenylketonuria nyebabake pelanggaran oksidasi phenylalanine saka panganan, nyebabake konsentrasi ing getih (phenylalaninemia) lan cairan serebrospinal mundhak kanthi signifikan, lan tingkat tyrosine mudhun kanthi tepat. Kelenjaralan sing gedhe banget disingkirake kanthi ekskresi urin sing tambah metabolit - asam phenylpyruvic, phenylmilactic lan phenylacetic acid.
Gangguan metabolisme asam amino diiringi myelination serat saraf, penurunan pembentukan neurotransmitter (dopamin, serotonin, lan liya-liyane), mekanisme patogenetik sing bisa mundur saka mental lan penyakit mental.
Gejala phenylketonuria
Bayi anyar karo phenylketonuria ora duwe pratandha klinis penyakit. Biasane, kawujudan phenylketonuria ing bocah-bocah dumadi ing umur 2-6 wulan. Kanthi wiwitan dipakani, protein susu susu utawa penggantine wiwit ngetik awak bayi, sing ndadékaké perkembangan gejala pisanan sing ora spesifik - lesu, kadang kuatir lan eksitivitas hiper, regurgitasi, dystonia otot, sindrom konvulatif. Salah sawijining pratandha phenylketonuria sing awal yaiku mutahke sing terus-terusan, sing asring dikira salah minangka manifestasi stenosis pyloric.
Ing setengah taun kaping loro, lagane bocah ing pangembangan psikomotor katon nyata. Anak kasebut dadi kurang aktif, ora peduli, mandheg ngenali wong sing ditresnani, ora nyoba njagong lan ngadeg ing sikile. Komposisi saka urin lan kringet ora normal nyebabake ambu "mouse" khas (mambu jamur) sing ana ing awak. Asring ana kulit ing kulit, dermatitis, eczema, scleroderma.
Ing bocah-bocah sing duwe phenylketonuria sing ora nampa perawatan, mikrocephaly, prognathia, mengko (sawise 1.5 taun), enamel hypoplasia dideteksi. Kelewatan pangembangan wicara kacathet, lan nganti taun-taun oligofrenia jero (idiocy) lan ora ana tuturan sing meh lengkap.
Bocah-bocah sing duwe phenylketonuria duwe struktural sing sithik, asring cacat jantung kongenital, disfungsi autonomi (kringet, akrocyanosis, hipotensi arteri), lan ngalami sembelit. Karakteristik fenotypic anak sing ngalami phenylketonuria kalebu kulit, mata lan rambut sing entheng. Bocah cilik karo phenylketonuria ditondoi kanthi nuduhake "buntut" (anggota ndhuwur lan ngisor mbengkongake sendi), tremor tangan, wobbly, mincing gait, lan hyperkinesis.
Manifestasi klinis phenylketonuria jinis II ditondoi kanthi tingkat retardasi mental sing abot, tambah jengkel, kejang, tetraparesis spastic, lan hiperlexlex tendon. Penyebaran penyakit kasebut bisa nyebabake pati bocah sing umur 2 nganti 3 taun.
Nalika jinis phenylketonuri III nggawe triad tandha: mikrofephaly, oligofrenia, tetraparesis spastic.
Diagnosis phenylketonuria
Saiki diagnosis diagnosis phenylketonuria (uga galaksi, hipotiroidisme kongenital, sindrom adrenogenital lan fibrosis sista) minangka bagean saka program screening neonatal kanggo kabeh bayi sing anyar.
Tes skripsi ditindakake sajrone umur 3 dina kanthi umur lengkap lan 7 dina urip bayi durung wayahe kanthi njupuk conto getih kapiler ing wangun kertas khusus. Yen hiperklikemiaemia ditemokake, luwih saka 2,2 mg% bocah kasebut disebut genetika pediatrik kanggo ditliti maneh.
Kanggo konfirmasi diagnosis phenylketonuria, konsentrasi phenylalanine lan tyrosine ing getih dicenthang, kegiatan enzim hepatik (phenylalanine hydroxylase) ditemtokake, pemeriksaan biokimia urin (tekad saka asam keton), metabolisme katekolamin ing urin, lan uga ditindakake.
Cacat genetik ing phenylketonuria bisa dideteksi sanajan meteng nalika diagnosis prenatal invasif saka janin (chorionbiopsy, amniosentesis, cordocentesis).
Diagnosis diferensial phenylketonuria ditindakake trauma kelahiran intrakranial nalika bayi, infeksi intrauterine, lan gangguan metabolik asam amino.
Perawatan Phenylketonuria
Faktor dhasar kanggo perawatan phenylketonuria yaiku panganan sing mbatesi asupan protein ing awak. Perawatan disaranake diwiwiti kanthi konsentrasi phenylalanine> 6 mg%. Campuran khusus wis dikembangake kanggo bayi - Afenilak, Lofenilak, kanggo bocah sing umur luwih saka 1 taun - Tetrafen, bebas Phenyl, luwih saka 8 taun - Maxamum-XP lan liya-liyane. Dasar dhasar yaiku panganan rendah protein - woh-wohan, sayuran, jus, hidrolisis protein lan campuran asam amino. . Ekspansi panganan bisa ditindakake sawise 18 taun sing ana hubungane karo toleransi phenylalanine. Sesuai karo hukum Rusia, panentu nutrisi medis kanggo wong sing nandhang phenylketonuria kudu gratis.
Pasien diwenehi asupan senyawa mineral, vitamin klompok B, lan liya-liyane, miturut indikasi - obatan nootropik, anticonvulsants. Ing terapi kompleks phenylketonuria, pijet umum, terapi latihan, lan akupunktur digunakake kanthi akeh.
Bocah-bocah sing nandhang phenylketonuria ana ing pengawasan ahli pediatrik lan neuropsychiatrist lokal, lan asring mbutuhake bantuan saka terapi wicara lan ahli patologi. Ngawasi ati-ati status neuropsikik bocah-bocah, pemantauan tingkat phenylalanine ing getih lan indikator elektronegephalogram perlu.
Bentuk phenylketonuria sing ora bisa ditrapake kanggo perawatan diet mbutuhake pelantikan hepatoprotectors, anticonvulsants, terapi gantian karo levodopa, 5-hydroxytryptophan.
Prediksi lan nyegah phenylketonuria
Nindakake screening massa kanggo phenylketonuria ing wektu neonatal ngidini sampeyan ngatur terapi diet awal lan nyegah karusakan serebral sing abot, fungsi ati mboten saget. Kanthi janjian awal kanggo diet penghapusan kanggo phenylketonuria klasik, ramalan kanggo pangembangan bocah-bocah apik. Kanthi perawatan pungkasan, ramalan kanggo pangembangan mental ora sehat.
Nyegah komplikasi phenylketonuria kasusun ing screening massa saka bayi, resep awal lan pematuhan jangka panjang karo nutrisi panganan.
Kanggo ngevaluasi risiko nglairake anak sing duwe phenylketonuria, penyuluhan genetik awal kudu diwenehake kanggo pasangan sing wis duwe anak sing lara, ana ing hubungan sing setuju, lan duwe sederek karo penyakit iki. Wanita sing duwe phenylketonuria sing ngrancang meteng kudu ngetutake diet sing ketat sadurunge angen-angen lan sajrone meteng kanggo ngilangi tingkat phenylalanine lan metabolit lan pangembangan janin sing sehat genetically. Resiko duwe anak karo phenylketonuria ing wong tuwa sing nggawa gawat cacat yaiku 1: 4.






-
Gula getih 6
Gula getih 6,5 unit, apike kanggo panganan sing paling disenengi lan nyuda diabetes? Kanggo perawatan sendi, para pamaca wis nggunakake DiabeNot. Kanggo ndeleng popularitas produk iki, kita mutusake kanggo menehi perhatian. ... -

-

-